Traitement et évolution :
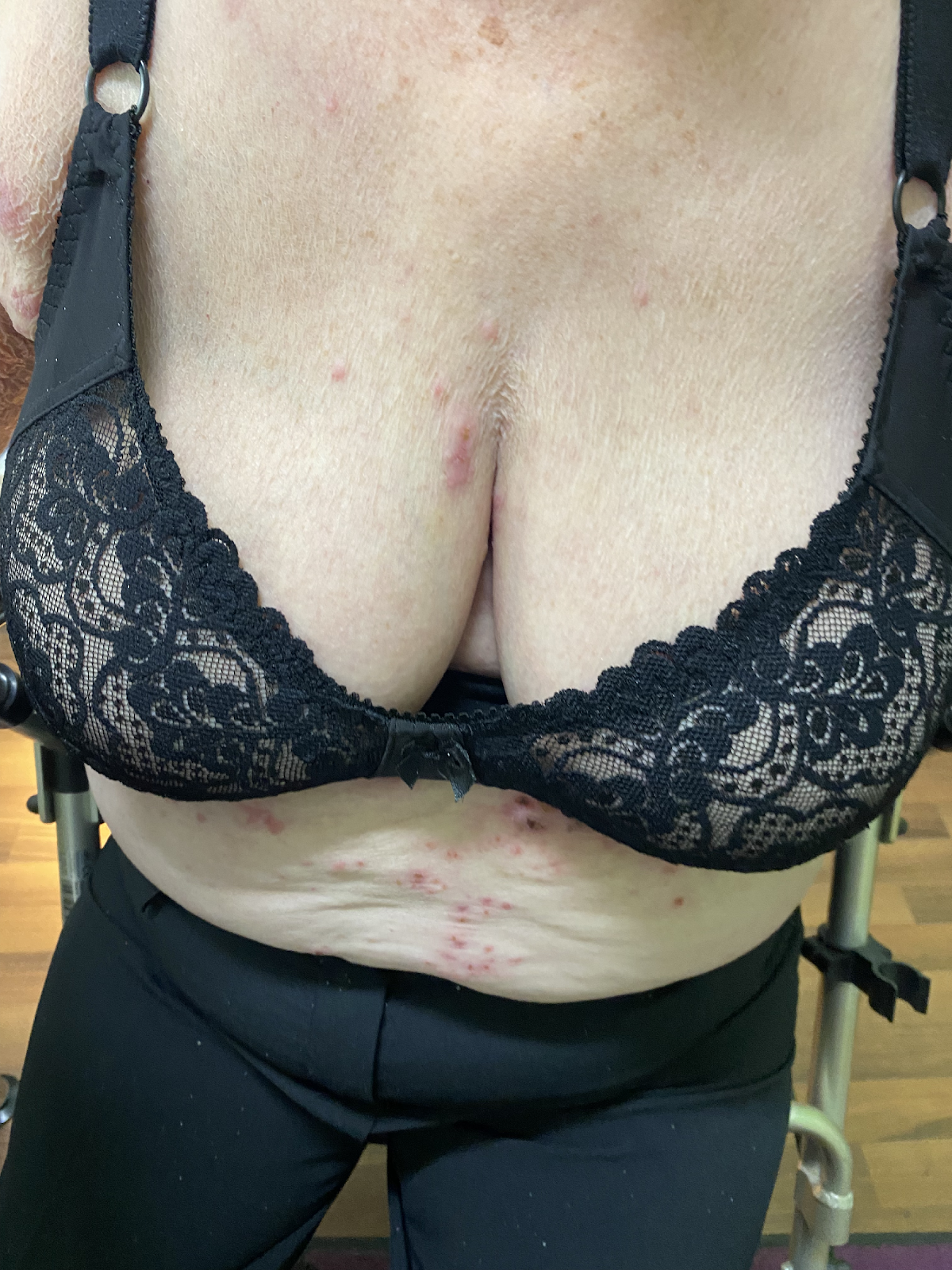
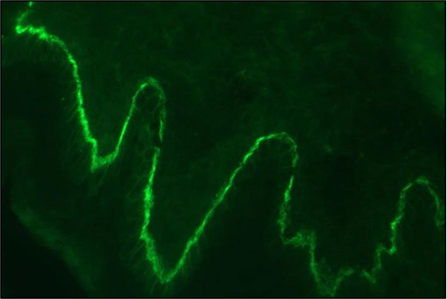
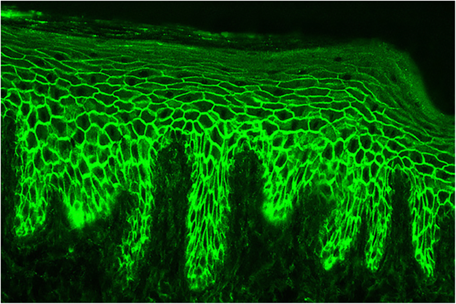
Les grandes bulles de tension séreuses tendues ainsi que les lésions cutanées (confirmées par l'anamnèse et existant depuis longtemps sur le bas du tronc et les extrémités proximales de la patiente âgée) étaient déjà fortement évocatrices d'une pemphigoïde bulleuse (PB), avant même que les résultats du diagnostic paraclinique et biopsique ne soient disponibles. Rétrospectivement, les lésions cutanées précédant l'éruption vésiculaire sont caractéristiques. Elles ont généralement un aspect eczémateux et urticarien et peuvent, en tant qu'« érythème prémonitoire », précéder la formation des bulles de plusieurs mois à plusieurs années. Le diagnostic de suspicion de PB a été confirmé par des sérologies auto-immunes, par l'histologie et par l'immunofluorescence directe.
Un traitement stéroïdien systémique par prednisolone 1mg/kg a été instauré avant même la réception des résultats. Une crème à base de bétaméthasone a également été appliquée localement. L'apaisement du prurit a été réalisé avec de la lévocétirizine p.o. et de la doxépine p.o. Le traitement a permis une régression relativement rapide des lésions cutanées aiguës et l'arrêt de la formation de nouvelles cloques. Sur une période de 14 jours, la dose de stéroïdes a pu être réduite à 10mg/d p.o.. En outre, le traitement antidiabétique par sitagliptine a été arrêté et remplacé par une insuline à action prolongée.
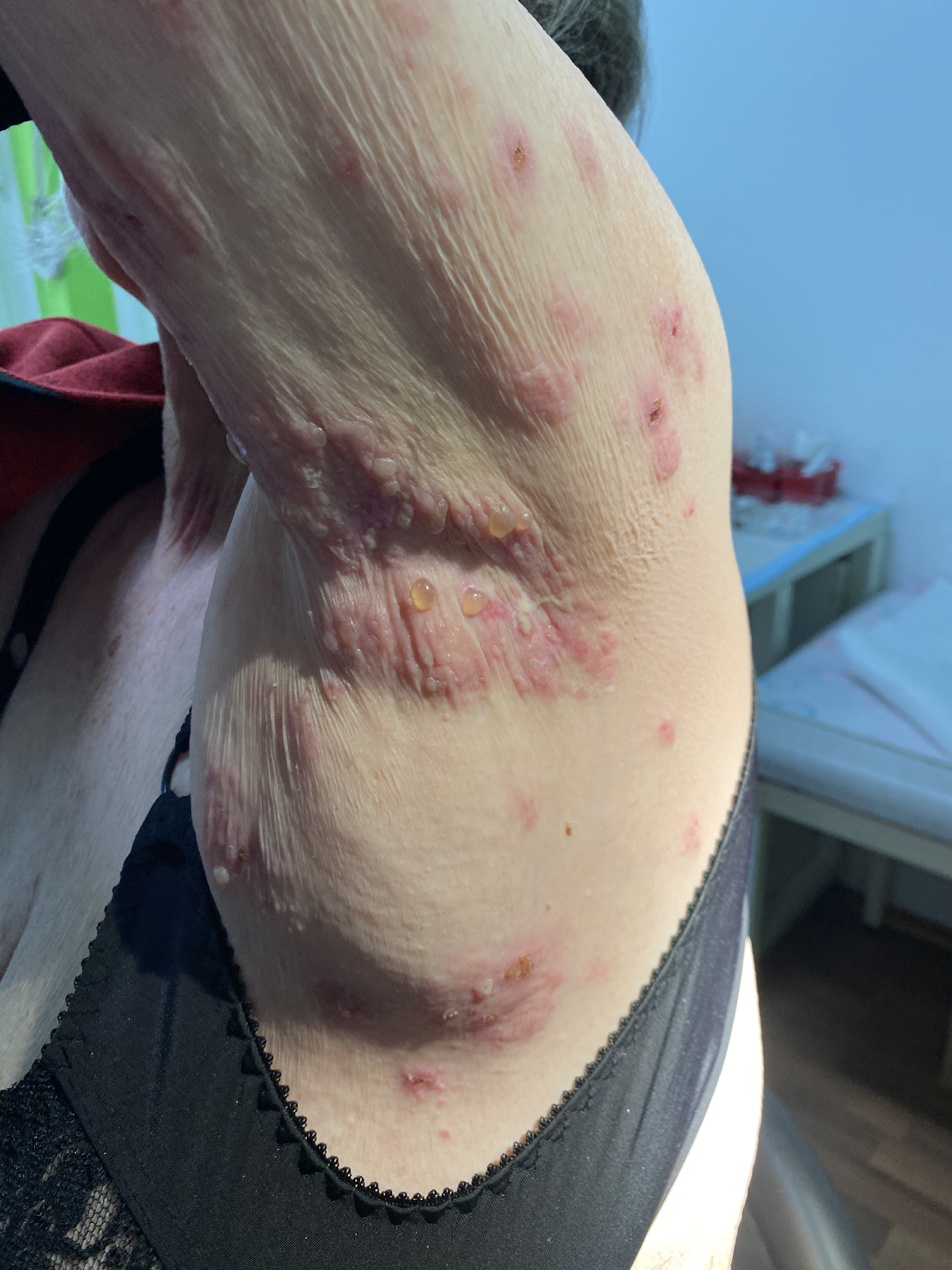
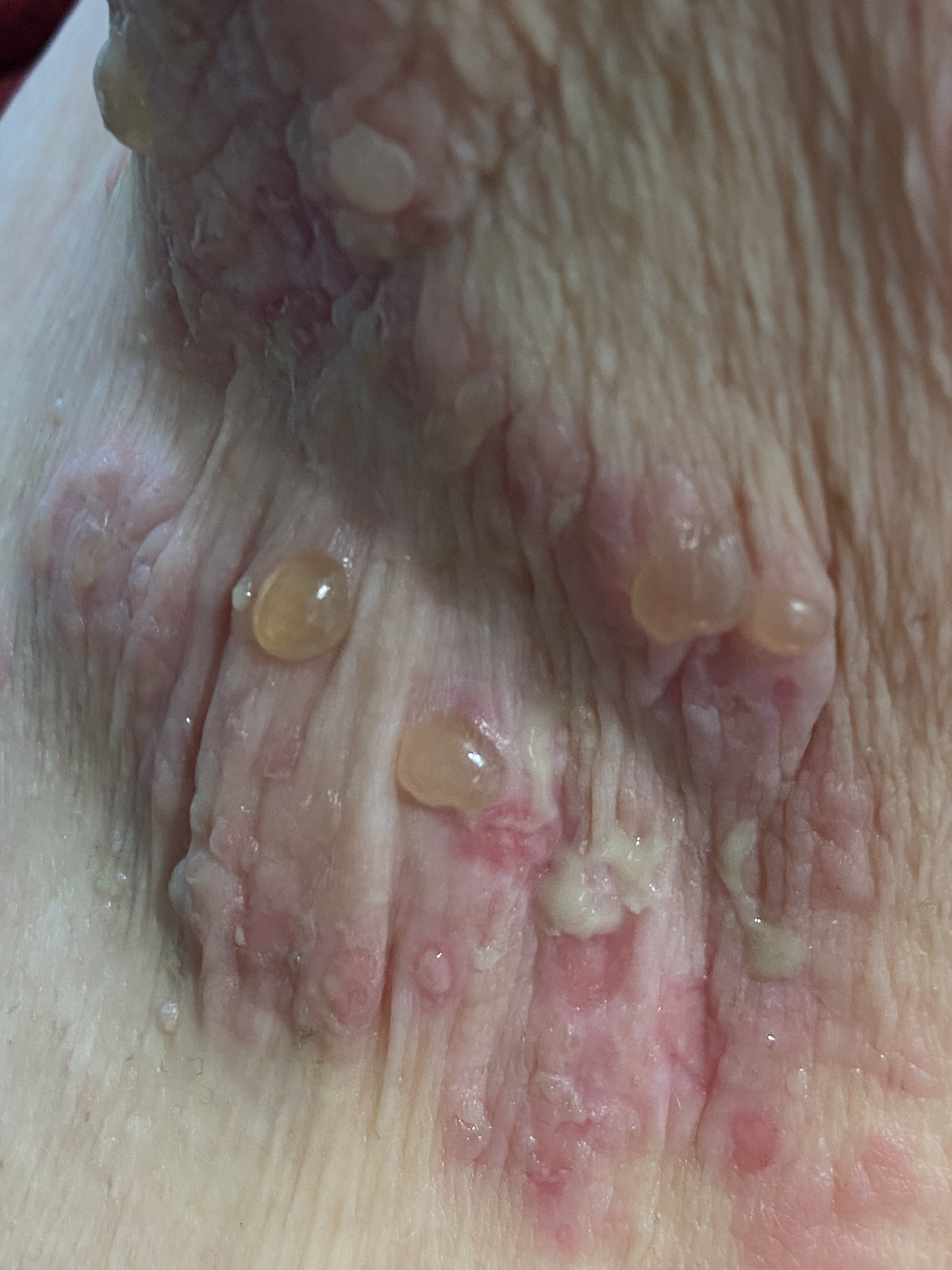
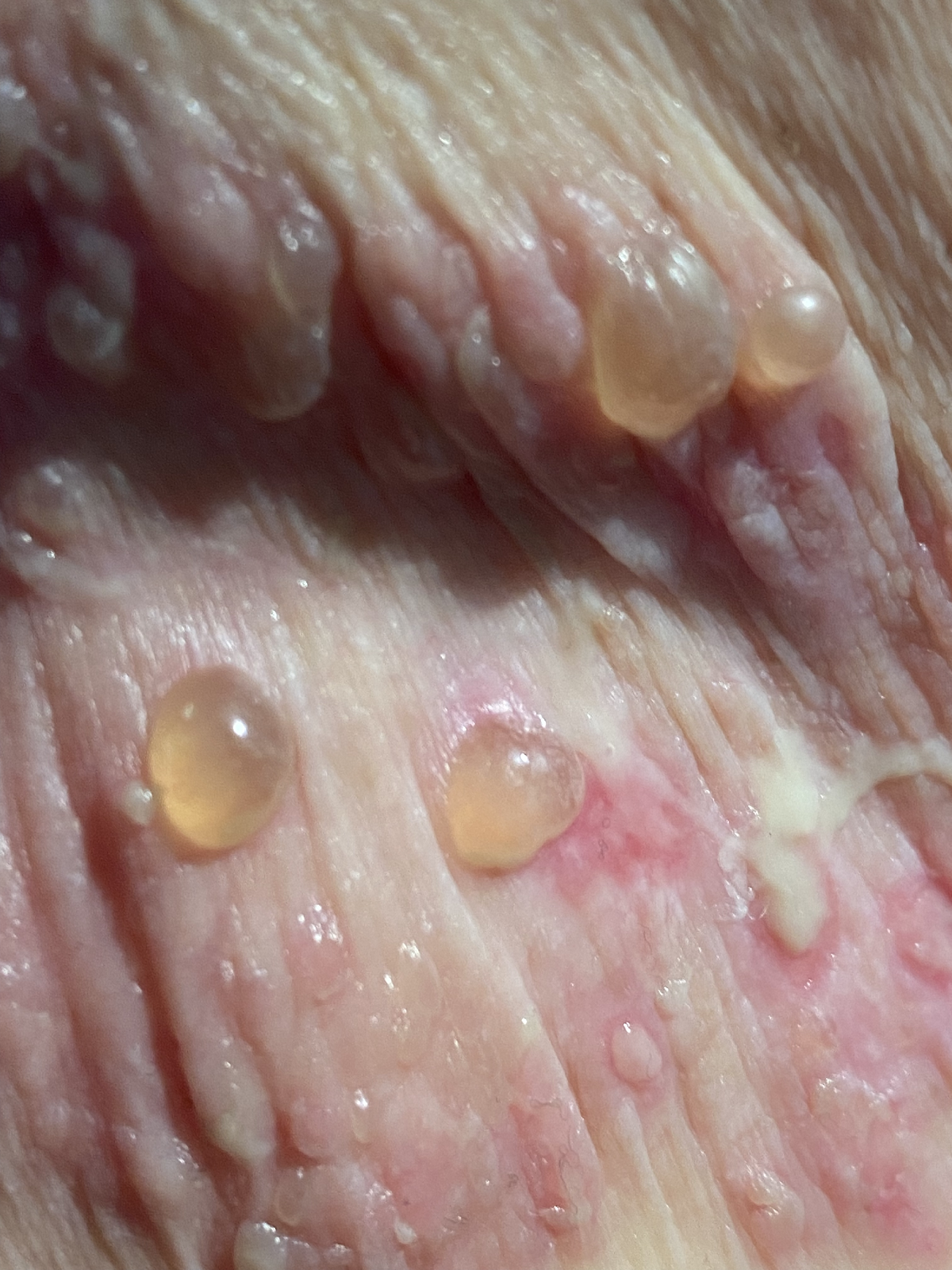
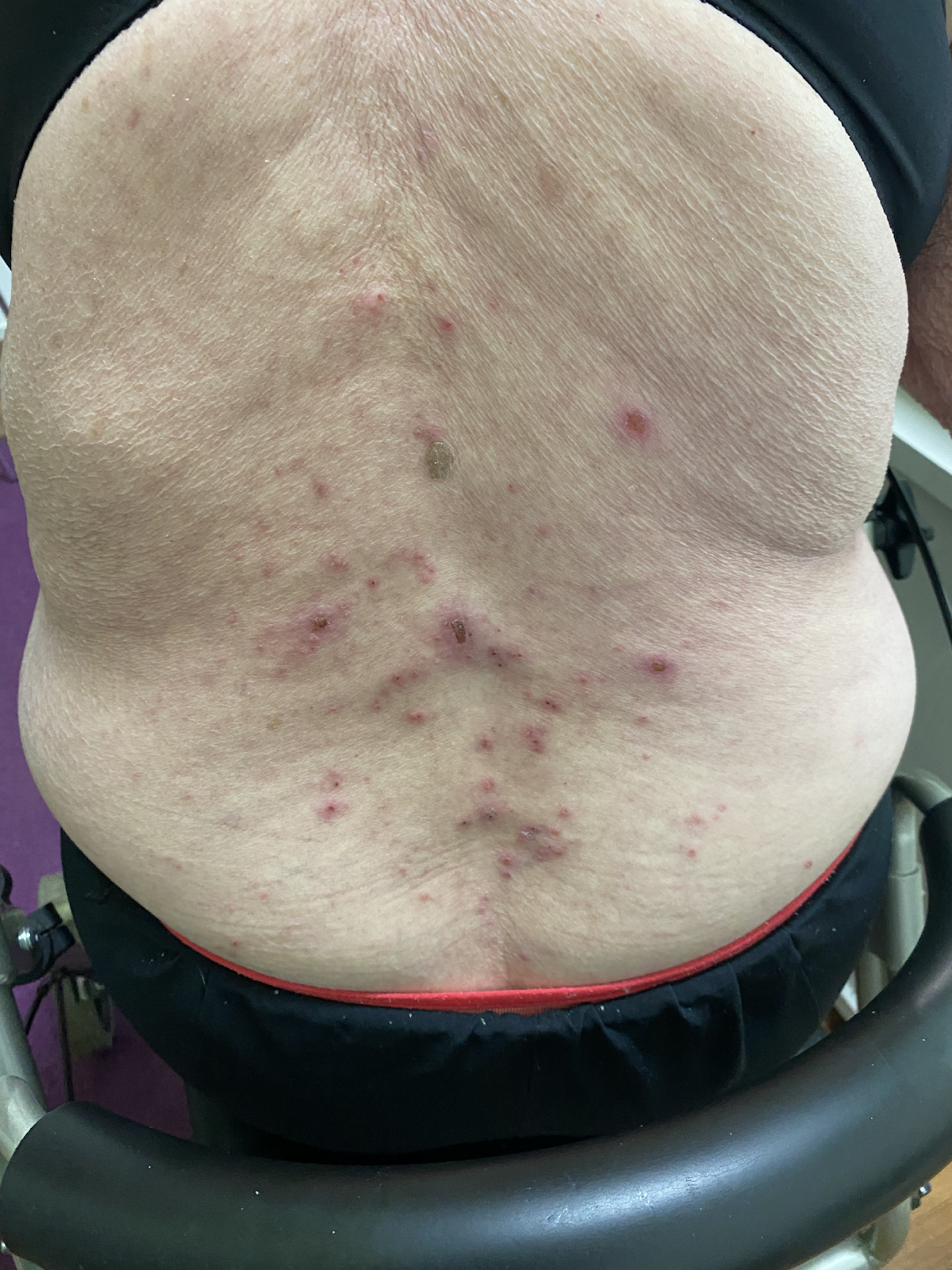
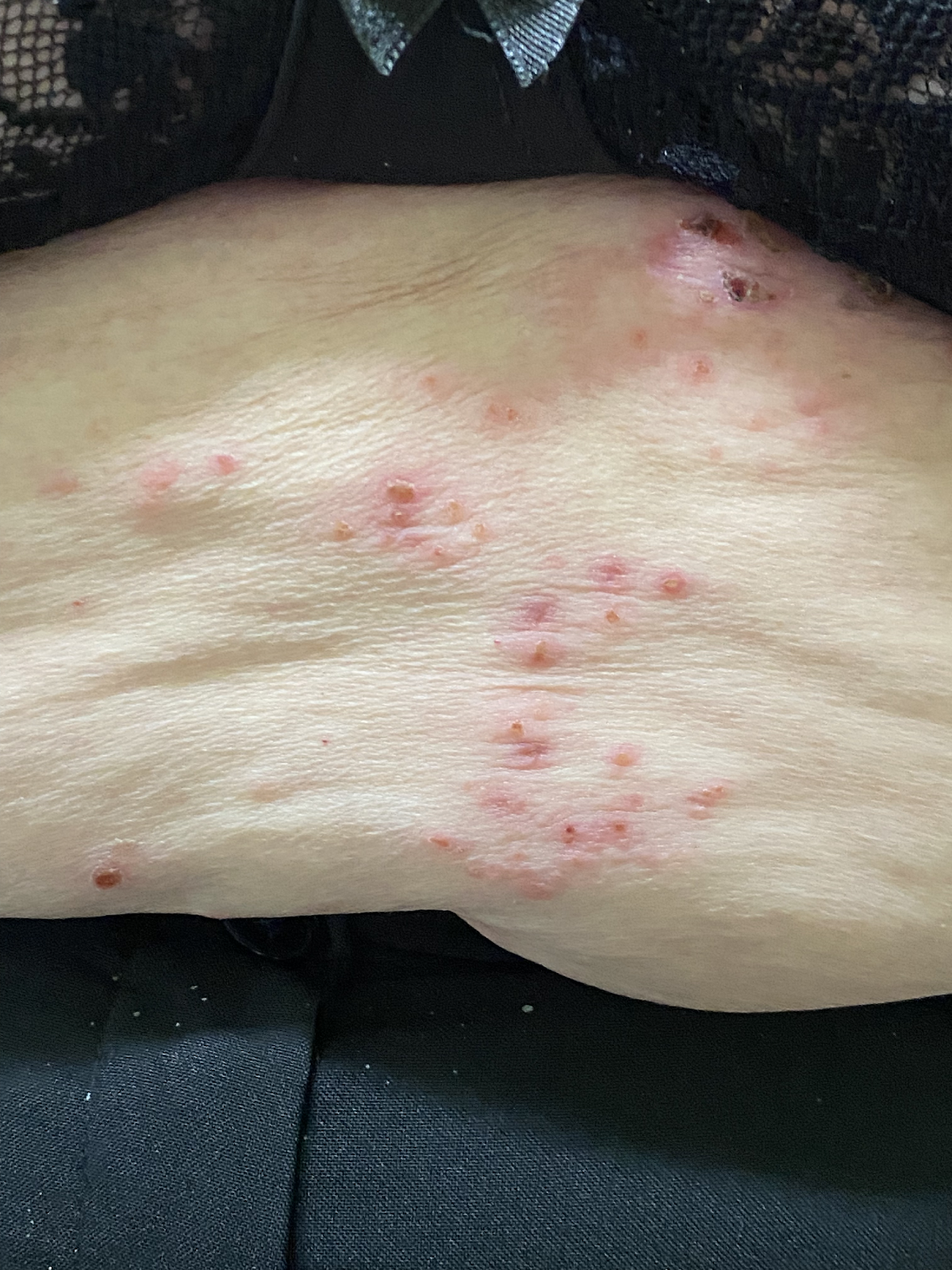
Les complexes d'auto-anticorps dirigés contre les protéines structurelles hémidesmosomales de la membrane basale, BP180 (BPAg2) et BP230 (BpAg1), provoquent leur détachement par activation du complément et migration des leucocytes. L'ensemble de l'épiderme constitue ainsi le toit de la bulle et il en résulte des bulles très stables, appelées bulles tendues. Les bulles sont le plus souvent séreuses, mais elles peuvent être hémorragiques, notamment chez les patients sous anticoagulants oraux (Image 3).
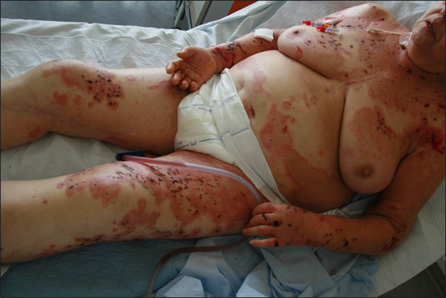
a

b
Image 3 : Patiente de 75 ans présentant une pemphigoïde bulleuse étendue. Ici, outre les bulles tendues en partie hémorragiques, on voit également les lésions cutanées en partie eczémateuses et urticariennes qui précèdent la formation des bulles (a,b).
Plus rarement, dans environ 20% des cas, les muqueuses, principalement orales, sont touchées. Il convient toutefois de distinguer la pemphigoïde muqueuse cicatricielle en tant qu'entité distincte, qui présente un schéma de CA hétérogène. Dans ce cas, l'atteinte des muqueuses est prédominante, et en particulier, l'atteinte conjonctivale conduit à la formation de synblépharon et d'ectropion irréversibles.
Les maladies bulleuses auto immunes (MBAI) représentent un groupe d'environ 20 pathologies hétérogènes caractérisées par des auto-anticorps dirigés contre des protéines structurelles de la peau et/ou des muqueuses. Selon le lieu d'attaque des auto-anticorps, on distingue les MBAI sous-épidermiques (p. ex. pemphigoïde bulleux (PB)) des MBAI intra-épidermiques (p. ex. pemphigus vulgaire). La pemphigoïde bulleuse peut se manifester sous forme de paranéoplasie (15%), bien qu'un lien pertinent avec une tumeur maligne ait été plutôt surestimé dans les années passées2. La plupart des cas sont idiopathiques, mais certains médicaments comme la pénicilline, le salazosulfapyridine, le diazépam ou le furosémide peuvent déclencher un PB.
Un lien avec la prise d'autres médicaments tels que la spironolactone, les phénothiazines ou les inhibiteurs de points de contrôle immunitaire a également été suggéré.3 Récemment, des cas de pemphigoïde bulleux déclenchés par les inhibiteurs de la DPP-4, tels que la sitagliptine, se sont multipliés.4 Les données de pharmacovigilance indiquent en effet un lien avec la prise d'inhibiteurs de la DPP-4 dans une partie des cas et il est recommandé de les remplacer par d'autres antidiabétiques en cas de diagnostic de BP. Les données d'études indiquent que ce lien existe surtout chez les patients de sexe masculin.5
Dans le cas présent, le lien temporel entre l'introduction du traitement par sitagliptine et le début des symptômes cutanés laissait supposer que l'inhibiteur de la DPP-4 en était la cause. Après l'arrêt de la sitagliptine, une guérison durable de la dermatose auto-immune a pu être obtenue sous une dose d'entretien prolongée de prednisolone de 5mg/j.
En France, le traitement de la pemphigoïde bulleuse suit une approche progressive, avec des traitements adaptés à la gravité et à l'évolution de la maladie.
Les options principales incluent6,7 :
- Des corticostéroïdes topiques : le clobétasol est recommandé comme traitement initial pour les cas modérés. En traitement systémique, on utilise souvent la prednisone (0,5 mg/kg/jour) pour les cas plus avancés.
- Des antibiotiques : la doxycycline est également utilisée pour contrôler les symptômes et améliorer la tolérance sur le long terme.
- Des immunosuppresseurs : pour les formes persistantes ou réfractaires, des immunosuppresseurs comme l'azathioprine ou le mycophénolate mofétil peuvent être prescrits en fonction de la réponse du patient et de l'atteinte des muqueuses.
- Le rituximab : Pour les formes sévères et résistantes,des traitements plus spécifiques comme le rituximab (antocorps anti-CD20) est recommandé dans les cas réfractaires, et parfois même pour des formes modérées.
En cas de PB prémonitoires, le diagnostic différentiel doit surtout être posé avec le prurigo, l'urticaire, la scabiose, mais aussi avec les maladies lymphoprolifératives. En cas de formation localisée de bulles, il faut distinguer les causes mécaniques ou traumatiques, l'impétigo contagiosa, l'érysipèle bulleux, l'herpès zoster, la dermatite de contact bulleuse, la réaction (bulleuse) à l'ictus, la réaction médicamenteuse bulleuse généralisée et bien sûr d'autres dermatoses bulleuses auto-immunes.2
Conclusion : traiter efficacement la pemphigoïde bulleuse avec des thérapies modernes
La pemphigoïde bulleuse est la dermatose auto-immune la plus fréquente chez les personnes âgées. Elle se caractérise par des bulles de tension stables sur le tronc et les extrémités proximales. Le début généralement prolongé sous forme d'« érythème prémonitoire » explique le diagnostic souvent tardif. La pemphigoïde bulleuse est le plus souvent idiopathique, mais elle peut aussi être paranéoplasique ou déclenchée par des médicaments. Dans ce cas, les observations liées à l'administration d'antagonistes de la DPP-4 sont de plus en plus fréquentes. Le traitement de choix en cas de poussée aiguë est le choc stéroïdien systémique. En cas d'évolution chronique, les immunosuppresseurs classiques comme l'azathioprine et le cycloiphosphamide, mais aussi les agents biologiques modernes comme le rituximab, sont recommandés. Lors du diagnostic de la pemphigoïde bulleuse, il convient de délimiter un large diagnostic différentiel.
Sources:
- Deotto ML, Spiller A, Sernicola A et al. Bullous pemphigoid: An immune disorder related to aging (Review). Exp Ther Med 2022; 23: 50. Persson MSM, Begum N, Grainge MJ et al. The global incidence of bullous pemphigoid: a systematic review and meta-analysis. Br J Dermatol 2021
- Czaika et al: Kurzlehrbuch Dermatologie. Autoimmunerkrankungen der Haut. Thieme-Verlag, 3.Auf. 2ß23, S161ff.
- 14 Geisler AN, Phillips GS, Barrios DM et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol 2020; 83: 1255–1268. doi:10.1016/j.jaad.2020.03.132
- Mohme S et al. Blasenbildende Autoimmundermatosen - Klinik, Diagnostik und neue Therapieansätze. Akt Rheumatol 2022; 47: 333–343 © 2022. Thieme
- Lee SG et al. Association of Dipeptidyl Peptidase 4 Inhibitor Use With Risk of Bullous Pemphigoid in Patients With Diabetes. JAMA Dermatol 2019;155(2):172-177
- Haute Autorité de Santé. Pemphigoïde bulleuse, Guide maladie chronique. 5 juin 2020
- Cochrane Skin. Interventions for bullous pemphigoid. 7 septembre 2023